
 International
International
News release
From:
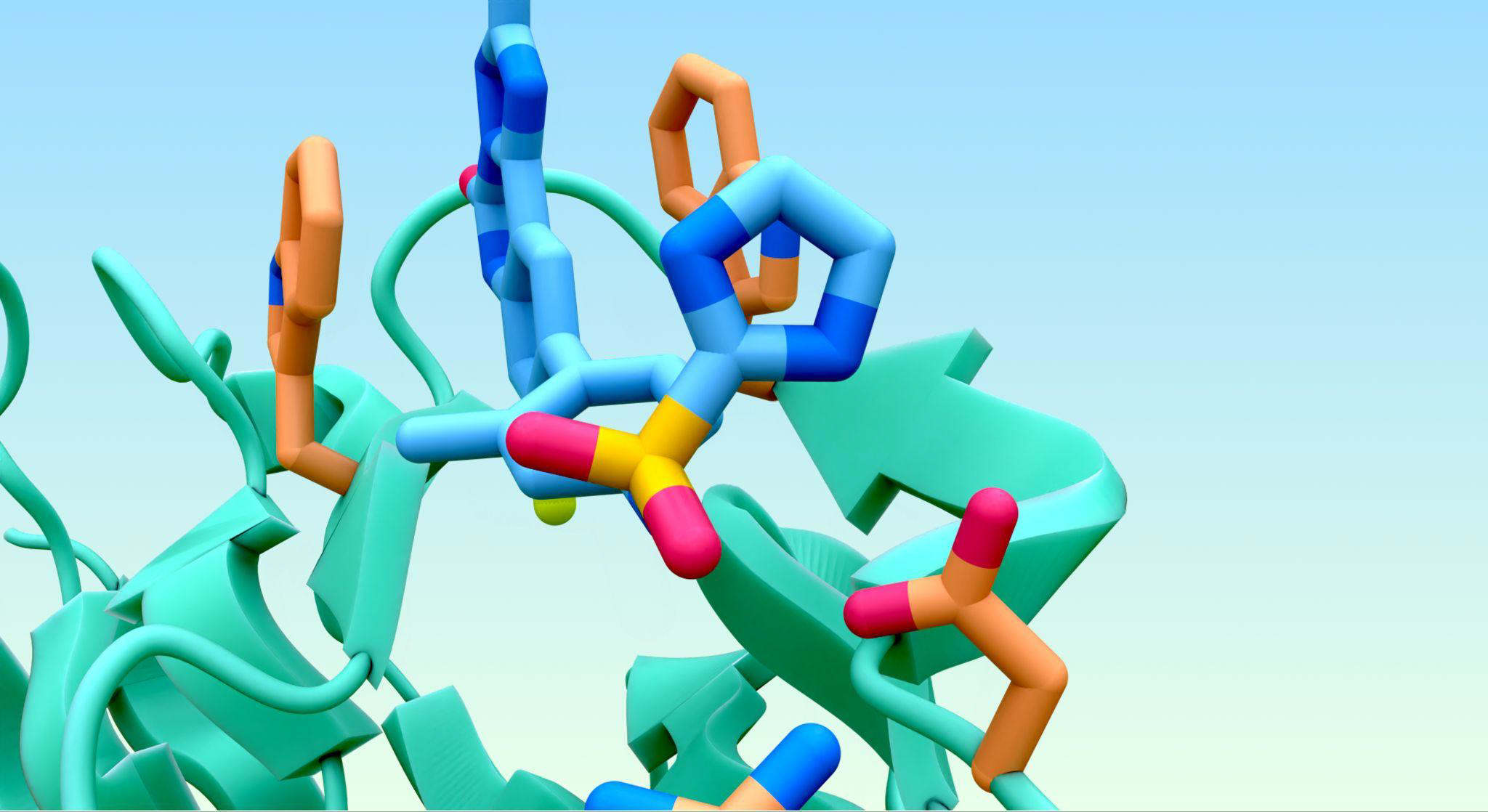
Structural biology: AlphaFold 3 widens and improves structure prediction accuracy for protein–molecule interactions *PRESS BRIEFING*
The ability of AlphaFold 3 to produce highly accurate predictions of the structures of proteins interacting with other biological molecules is reported in Nature this week. AlphaFold 3, the latest iteration of the artificial intelligence model created by researchers at Google DeepMind and Isomorphic Labs, demonstrates substantially better accuracy over previous specialist tools. This new model can predict the structures of complexes containing nearly all molecule types within the Protein Data Bank. The ability to computationally determine complex interactions between proteins and other molecules will expand our understanding of biological processes and could facilitate drug development.
First released in 2020, AlphaFold and its later iteration, AlphaFold 2, enabled prediction of the 3D structure of a protein from its sequence of amino acids (the building blocks of proteins). Subsequently, AlphaFold-Multimer facilitated prediction of protein–protein complexes. Broadening the range of complexes whose structure a single deep learning model can predict has been challenging, owing to the vast differences in specific interaction types.
Substantial improvements to the AlphaFold 2 model’s deep learning architecture and training system have now made it possible to more accurately predict the structure of a wide range of biomolecular systems in a unified framework, John Jumper and colleagues report. AlphaFold 3 can predict complexes of proteins with other proteins, nucleic acids, small molecules, ions and modified protein residues, as well as antibody–antigen interactions. Its accuracy significantly exceeds that of current prediction tools, including AlphaFold-Multimer.
The authors note some limitations, such as incorrect chirality (a symmetry property) occurring in around 4.4% of structures, or hallucinations resulting in a reduction in the appearance of ribbons (a common protein secondary structure element). They add that further improvements in modelling accuracy would require the generation of a large set of predictions and ranking of the resulting structures, which would incur an additional computational cost.
Attachments
Note: Not all attachments are visible to the general public. Research URLs will go live after the embargo ends.


