
 International
International
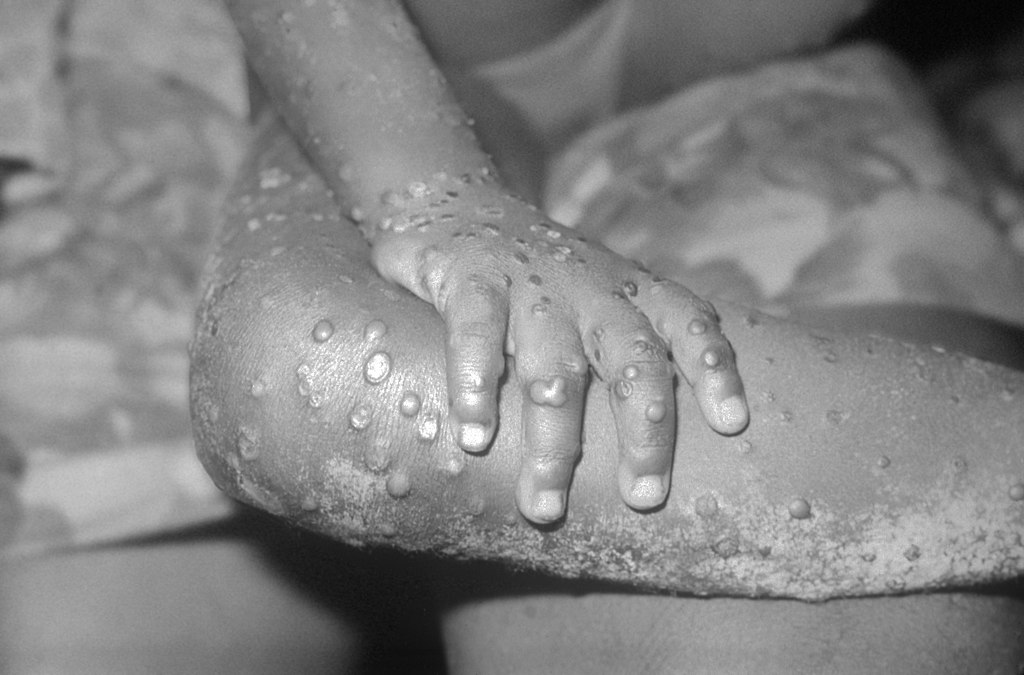
News release
From:
Genomic data sheds light on the evolutionary trajectory of the monkeypox outbreak
The strain associated with the current monkeypox virus outbreak identified in May 2022 is a well-defined divergent branch from the monkeypox viruses of a 2018–2019 outbreak in an endemic country that probably represents recent evolutionary changes. This study, published in Nature Medicine, also highlights ongoing evolution during human-to-human transmission, which may explain increased transmission of this strain.
Monkeypox is a rare infectious disease that spreads between species, including from animals to humans, caused by the monkeypox virus (MPXV) of the Orthopoxvirus genus (which also includes smallpox virus). Monkeypox is endemic to West and Central African countries, and the infrequent reports of cases outside those regions are associated with importation from those countries. The first multi-country outbreak of monkeypox was identified in May 2022, with more than 2,500 confirmed cases worldwide, as of 18 June.
To investigate how the 2022 outbreak began, João Paulo Gomes and colleagues reconstructed genome sequences of the MPXV associated with the outbreak. Their analysis revealed that this MXPV belongs to the MPXV clade 3 and that the ongoing outbreak most likely has a single origin. The 2022 MPXV diverges from the related 2018–2019 viruses by about 50 single-nucleotide polymorphisms, or genetic variations — far more than expected for orthopoxviruses. Such a divergent branch may represent ongoing accelerated evolution, according to the authors. Further analysis revealed the first signs of ongoing evolution — 15 single-nucleotide polymorphisms, minor variants and gene deletion — during human-to-human transmission within the ongoing outbreak.
Although further research is needed, these findings help elucidate the evolutionary trajectory of the 2022 MPXV outbreak strain by providing data on potential mechanisms underlying viral evolution and possible viral gene targets of human adaptation.
Attachments
Note: Not all attachments are visible to the general public. Research URLs will go live after the embargo ends.


